This chapter describes the Car-Parrinello molecular dynamics simulations which
have been performed to investigate the solvation effects on the
prototype S
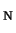
2 reaction between Cl

and CH
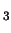
Cl.
The free energy barrier for this reaction in water was calculated using
constrained dynamics at a constant temperature of
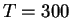
K and a constant volume.
Calculations on the isolated system (reaction in the gas phase at zero temperature) were
performed for reference purposes.
Qualitatively the calculations confirm that the double-well free energy profile
of the reaction in the gas phase is converted into a single barrier by solvation,
and that the height of the barrier increases significantly.
Quantitatively, there are two error sources. At the electronic structure side the
Becke-Perdew functional underestimates the barrier height by 8 kcal/mol. At the
dynamics side there is a "hysteresis" effect - too slow adaptation of the solvent
structure to changes in the reaction coordinate - yielding an estimated error of
3 kcal/mol in the free energy barrier height. After correction for these errors
the calculated value of the free energy barrier is 27 kcal/mol.
Considering the accuracy of the solvent-solvent and solvent-reactant interactions
of ca. 1 kcal/mol, this compares very well with the experimental estimate
of 26.6 kcal/mol. This indicates that the
ab initio (DFT) MD very well captures the differential energetic as well as entropic
effects of the solvation when going from the (solvated) reactants to the initial
ion-dipole complex to the transition state.