To study the solvation effects on the reaction between CH![]() Cl with
Cl
Cl with
Cl![]() , we will first consider the gas phase reaction. Starting with
separated reactants, the reaction energy
, we will first consider the gas phase reaction. Starting with
separated reactants, the reaction energy ![]() decreases as the attacking chloride anion approaches the dipolar
CH
decreases as the attacking chloride anion approaches the dipolar
CH![]() Cl from the carbon side to form a reaction complex, with
complexation energy
Cl from the carbon side to form a reaction complex, with
complexation energy
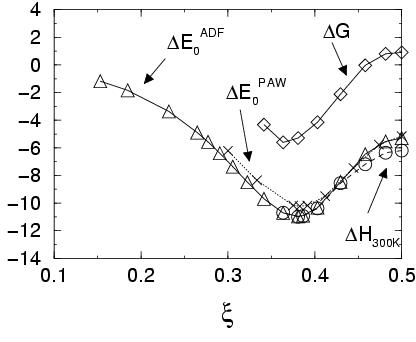
![]() (see figure 3.4).
Moving along to the product side,
(see figure 3.4).
Moving along to the product side,
![]() increases until the reactants arrive at the
increases until the reactants arrive at the ![]() symmetric transition state, with energy
symmetric transition state, with energy
![]() . The
product side of the profile is symmetric to the reactant side. The
numerous attempts to estimate
. The
product side of the profile is symmetric to the reactant side. The
numerous attempts to estimate
![]() and
and
![]() have resulted, especially for the latter, in a
variety of values for these quantities. A selection of these energies
and the corresponding geometries found in the literature, as well as our
own results are compiled in table 3.6. For the
ion-dipole reaction complex energy, the best ab initio number is
probably given by the G2 calculation of Glukhovtsev et al.:
have resulted, especially for the latter, in a
variety of values for these quantities. A selection of these energies
and the corresponding geometries found in the literature, as well as our
own results are compiled in table 3.6. For the
ion-dipole reaction complex energy, the best ab initio number is
probably given by the G2 calculation of Glukhovtsev et al.:
![]() kcal/mol.[58,123]
Our DFT results and the
ab initio and DFT results from literature are all within 1 kcal/mol
to this number. Also the experimental estimate by Larson and
McMahon agrees within their error estimate with this value. Only the CISD
(configurational interaction, including single and double excitations)
result by Vetter and Zülicke is about 2 kcal/mol too low.
kcal/mol.[58,123]
Our DFT results and the
ab initio and DFT results from literature are all within 1 kcal/mol
to this number. Also the experimental estimate by Larson and
McMahon agrees within their error estimate with this value. Only the CISD
(configurational interaction, including single and double excitations)
result by Vetter and Zülicke is about 2 kcal/mol too low.
|
|
||||||
| Reaction complex | Transition state | |||||
|
|
|
|
|
|
|
|
| [kcal/mol] | [Å] | [Å] | [kcal/mol] | [Å] | [kcal/mol] | |
|
|
||||||
|
|
||||||
| CP-PAW/BP | -10.39 | 1.91 | 3.01 | -5.32 | 2.37 | 5.1 |
|
|
||||||
| ADF-BP | -10.96 | 1.88 | 3.09 | -5.30 | 2.35 | 5.7 |
|
|
||||||
| DFT-BP |
-10.3 | 1.835 | 3.098 | -5.7 | 2.342 | 4.6 |
| MP2 |
-10.5 | 1.808 | 3.266 | 3.5 | 2.316 | 14.0 |
| MP4 |
-10.6 | 1.8 | 12.4 | |||
|
|
||||||
| MP2 |
4.01 | 2.28 | ||||
| CCSD(T) |
2.65 | 2.301 | ||||
|
|
||||||
| G2 |
-10.51 | 1.810 | 3.270 | 2.76 | 2.317 | 13.26 |
| DFT-B3LYP |
-9.72 | 1.858 | 3.180 | 1.1 | 2.371 | 10.8 |
|
|
||||||
| HF/CISD |
-8.7 | 1.823 | 3.384 | 8.7 | 2.408 | 17.5 |
|
|
||||||
| MP2 |
-9.66 | 1.808 | 3.267 | 7.68 | 2.316 | 17.34 |
| B3LYP |
-9.52 | -0.85 | 8.67 | |||
|
|
||||||
| Expt. | -8.6 |
1. |
13.2 |
|||
| -12.2 |
||||||
|
The results for the transition state energies, varying from -5.7
till 8.7 kcal/mol (see table 3.6) have lead to a number of
conflicting views. For instance, Streitwieser et al.
(ref StChAb97) concluded: ``the large differences in TS properties
between MP2 and B3LYP suggest that the latter may not always be
reliable for TS structures''. And Deng et al.
(ref debrzi94) concluded that ``the experimental data (for
![]() and
and
![]() ) seem to fall in
the region with the MP4 and NL-SCF (= DFT-BP) value as the upper and
lower bounds, respectively.'' The highest-level ab initio
result for
) seem to fall in
the region with the MP4 and NL-SCF (= DFT-BP) value as the upper and
lower bounds, respectively.'' The highest-level ab initio
result for
![]() is the CCSD(T) calculation by
Botschwina, equal to 2.65 kcal/mol. The G2 estimate by Glukhovtsev
et al. agrees very well with it, as well as the approximate
experimental result of 1.0
is the CCSD(T) calculation by
Botschwina, equal to 2.65 kcal/mol. The G2 estimate by Glukhovtsev
et al. agrees very well with it, as well as the approximate
experimental result of 1.0 ![]() 1.0 kcal/mol. This would imply that
the DFT-BP result of -5.7 kcal/mol by Deng is just too low, and
underestimates
1.0 kcal/mol. This would imply that
the DFT-BP result of -5.7 kcal/mol by Deng is just too low, and
underestimates
![]() significantly. Streitwiesers
MP2 result of 7.7 kcal/mol seems erroneous, compared to the MP2 results
of Botschwina (4.01 kcal/mol) and Deng et al. (3.5 kcal/mol).
The overestimation of the CISD energy (8.7
kcal/mol) is probably due to a combination of an inaccurate (HF)
geometry and a too small basis set (DZDP).
significantly. Streitwiesers
MP2 result of 7.7 kcal/mol seems erroneous, compared to the MP2 results
of Botschwina (4.01 kcal/mol) and Deng et al. (3.5 kcal/mol).
The overestimation of the CISD energy (8.7
kcal/mol) is probably due to a combination of an inaccurate (HF)
geometry and a too small basis set (DZDP).
There are indications that the too low transition state energy by
DFT-BP is systematic for structures with
a symmetrical three-center four-electron bond, such as the ![]() -bond
in Cl-C-Cl. For example, Gritsenko et al.[124]
investigated the very similar
-bond
in Cl-C-Cl. For example, Gritsenko et al.[124]
investigated the very similar
![]() transition state structure. They concluded that the delocalization of
the exchange hole over the three atoms in combination with a very
small non-dynamical correlation, is erroneously represented by the
exchange part of the GGA density functional, which introduces a
localized hole and thus a spurious non-dynamical correlation[124].
This is, of course, important to keep in
mind as we proceed to the S
transition state structure. They concluded that the delocalization of
the exchange hole over the three atoms in combination with a very
small non-dynamical correlation, is erroneously represented by the
exchange part of the GGA density functional, which introduces a
localized hole and thus a spurious non-dynamical correlation[124].
This is, of course, important to keep in
mind as we proceed to the S![]() 2 reaction in water
solution. Anticipating the results for the reaction in aqueous
solution, we may expect that the transition state in the solvated case
is underestimated by an amount in the order of 8 kcal/mol
because of the similarity in
the geometric and electronic structure of the reacting species. The
accuracy of the solvation effects should, in principle, be
in the order of 1 kcal/mol, as followed from the simulations in the
previous sections.
2 reaction in water
solution. Anticipating the results for the reaction in aqueous
solution, we may expect that the transition state in the solvated case
is underestimated by an amount in the order of 8 kcal/mol
because of the similarity in
the geometric and electronic structure of the reacting species. The
accuracy of the solvation effects should, in principle, be
in the order of 1 kcal/mol, as followed from the simulations in the
previous sections.
Figure 3.4 plots the reaction energy profile,
as well as the reaction enthalpy ![]() and the free energy
and the free energy
![]() . The latter two are calculated only between the reactant
complex and the transition state because of the failure of the smooth
change of vibrational contributions into translational and volume work
contributions for further separated reactants. The total correction
to the calculated energy
. The latter two are calculated only between the reactant
complex and the transition state because of the failure of the smooth
change of vibrational contributions into translational and volume work
contributions for further separated reactants. The total correction
to the calculated energy ![]() to obtain the enthalpy
to obtain the enthalpy ![]() at a temperature of
at a temperature of ![]() K (see eqn 3.3)
is very small for the ion-dipole complex. It
amounts to less than 0.1 kcal/mol for the equilibrium geometry, and
-0.9 kcal/mol for the transition state. This is in good agreement with
the estimates by Vetter and Zülicke (0.1 kcal/mol and
-0.5 kcal/mol, respectively). The formation of the ion-dipole complex from
infinitely separated reactants involves a large negative entropy
change, equal to
K (see eqn 3.3)
is very small for the ion-dipole complex. It
amounts to less than 0.1 kcal/mol for the equilibrium geometry, and
-0.9 kcal/mol for the transition state. This is in good agreement with
the estimates by Vetter and Zülicke (0.1 kcal/mol and
-0.5 kcal/mol, respectively). The formation of the ion-dipole complex from
infinitely separated reactants involves a large negative entropy
change, equal to
![]() kcal/mol. The entropy difference
(
kcal/mol. The entropy difference
(![]() )
of the transition state with respect to the free reactants is -7.1
kcal/mol, which means that the intrinsic free energy reaction barrier
is about 1.6 kcal/mol higher than the internal energy barrier.
)
of the transition state with respect to the free reactants is -7.1
kcal/mol, which means that the intrinsic free energy reaction barrier
is about 1.6 kcal/mol higher than the internal energy barrier.