The presence of solvent can have a major impact on the thermodynamics and kinetics of chemical reactions. Aspects such as electrostatic interactions and steric hindrance can play an important role in this respect. Also, chemical binding of solvent molecules to the reacting species may have a strong effect. A detailed microscopic picture of these effects is of fundamental interest and provides insight for possible improvements with respect to the rate, balance, and selectivity of chemical reactions.
Molecular simulation is an appropriate tool to obtain such a detailed picture. A high-level quantitative molecular simulation of a chemical reaction in solution requires the use of accurate quantum chemical methods to describe the change in chemical bonding in combination with a statistical mechanical description of the effect of the rearrangement of solvent molecules. The Car-Parrinello (CP) method[49] satisfies these two requirements in a computationally efficient manner. It combines Density Functional Theory (DFT) to describe the electronic structure with Molecular Dynamics (MD) to incorporate the motion of the molecules. The forces on the molecules that govern their motion are obtained directly from the electronic structure calculation. Therefore the method may be regarded as an ab initio (DFT) Molecular Dynamics (AIMD) simulation. Of particular importance for the present work is that AIMD has proven to be applicable to the study of the structure and dynamics of water[50,51,52,53] and simple chemical reactions in aqueous solution.[54,55]
The purpose of the present study is to gain microscopic insight in
the energetics, mechanism, and structural aspects of the solvation
effects on a prototype S![]() 2 reaction in water, with a
minimal level of approximation and without use of empirical data.
In particular we have investigated the effect of solvation in a dilute
aqueous HCl solution of the bimolecular nucleophilic substitution
(S
2 reaction in water, with a
minimal level of approximation and without use of empirical data.
In particular we have investigated the effect of solvation in a dilute
aqueous HCl solution of the bimolecular nucleophilic substitution
(S![]() 2) reaction,
2) reaction,
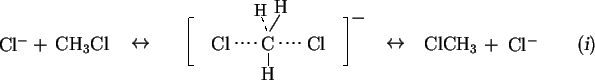
The S![]() 2 type of reaction is one of the most fundamental
processes in organic chemistry and has been
the subject of numerous theoretical
[56,57,58,59,60,61] and
experimental studies [62,63,64].
The energy profile for the gas phase reaction between methyl chloride and a
chloride anion is commonly believed to have a symmetrical double-well
shape with the two minima related to the ion-dipole complexes centered
around a maximum at the transition state. However, experimental
evidence shows that in aqueous solution the profile is unimodal and
the reaction rate much lower compared to that of the gas phase reaction. Using classical
Monte Carlo simulations, Chandrasekhar, Smith and Jorgensen[65] were the
first to compute the barrier for the Cl
2 type of reaction is one of the most fundamental
processes in organic chemistry and has been
the subject of numerous theoretical
[56,57,58,59,60,61] and
experimental studies [62,63,64].
The energy profile for the gas phase reaction between methyl chloride and a
chloride anion is commonly believed to have a symmetrical double-well
shape with the two minima related to the ion-dipole complexes centered
around a maximum at the transition state. However, experimental
evidence shows that in aqueous solution the profile is unimodal and
the reaction rate much lower compared to that of the gas phase reaction. Using classical
Monte Carlo simulations, Chandrasekhar, Smith and Jorgensen[65] were the
first to compute the barrier for the Cl![]() + CH
+ CH![]() Cl reaction in aqueous solution.
They developed model potentials for the interactions between the water molecules
and the solutes (Cl
Cl reaction in aqueous solution.
They developed model potentials for the interactions between the water molecules
and the solutes (Cl![]() and CH
and CH![]() Cl), and between water molecules, by fitting to
ab initio calculations. It is an advantage of the Car-Parrinello simulation
that these potentials are obtained directly. This is particularly important in a
case like the present one where construction of good model potentials is difficult
because of the changes in the charges on the attacking and leaving chloride ions,
and hence in their solvation, along the reaction coordinate.
Cl), and between water molecules, by fitting to
ab initio calculations. It is an advantage of the Car-Parrinello simulation
that these potentials are obtained directly. This is particularly important in a
case like the present one where construction of good model potentials is difficult
because of the changes in the charges on the attacking and leaving chloride ions,
and hence in their solvation, along the reaction coordinate.
The reaction studied in the present work is activated. The
experimental value for the activation energy is 27
kcal![]() mol
mol![]() .[66] A successful reaction is therefore
a rare event and outside the time scale accessible to ab initio
Molecular Dynamics. To study such reactions by MD a reactive encounter
must be forced on the system by some form of microscopic control of a
suitable reaction coordinate. This is achieved by using the method of
constraint that allows for the evaluation of the free-energy profile
along the reaction path.[16]
.[66] A successful reaction is therefore
a rare event and outside the time scale accessible to ab initio
Molecular Dynamics. To study such reactions by MD a reactive encounter
must be forced on the system by some form of microscopic control of a
suitable reaction coordinate. This is achieved by using the method of
constraint that allows for the evaluation of the free-energy profile
along the reaction path.[16]
This paper is organized as follows: In section 3.2, we
describe the computational details of the DFT and MD method, the
method of constrained dynamics to calculate the reaction free energy,
and the relevant parameters for the gas phase and aqueous solution
calculations. Section 3.3 starts with the results of some
preparatory simulations that serve to assess the accuracy of the
methods used. The simulation results of the S![]() 2 reaction in
gas phase and in aqueous solution are presented in sub-sections
3.3.3 and 3.3.4,
respectively. In section 3.4
a discussion of our results for the solvation effect
on the reaction barrier is given and we end with conclusions in
section 3.5.
2 reaction in
gas phase and in aqueous solution are presented in sub-sections
3.3.3 and 3.3.4,
respectively. In section 3.4
a discussion of our results for the solvation effect
on the reaction barrier is given and we end with conclusions in
section 3.5.