The pathway relaxation from an artificially constructed reaction pathway
to a pathway without the memory of the initial construction, was obtained
in two steps (for both sequences A and B).
The first step took place in the first pathway generation,
namely by taking completely random atomic momenta at
the starting point (which was the transition state configuration of
the initial reaction pathway). The second part of the relaxation consisted
of nine more sequential pathway generations, in which we observed
(a) increasingly faster terminations of the leaving OH. radical
due to shorter lifetimes
![]() and
(b) a shift from the direct rebound mechanism and the long-wire
two-step mechanism to the short-wire two-step mechanism.
and
(b) a shift from the direct rebound mechanism and the long-wire
two-step mechanism to the short-wire two-step mechanism.
|
These changes along the two sequences of pathways can be
understood as the result of the relaxation of the solvent environment
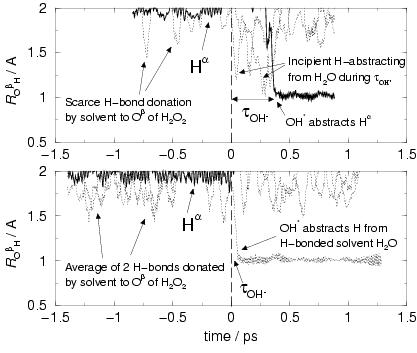
of the reactants. As an illustration, figure 7.6
shows the hydrogen-oxygen distances
![]() as a function
of time for the solvent water hydrogens within a radius of 2 Å
of the H
as a function
of time for the solvent water hydrogens within a radius of 2 Å
of the H![]() O
O![]() oxygen, O
oxygen, O![]() , for the initial unrelaxed
reaction pathway (upper graph) and the last pathway (no. 10) of sequence A
(lower graph). The moment that the hydrogen peroxide O-O distance
equaled
, for the initial unrelaxed
reaction pathway (upper graph) and the last pathway (no. 10) of sequence A
(lower graph). The moment that the hydrogen peroxide O-O distance
equaled
![]() Å, which marks the moment of coordination
and dissociation of the hydrogen peroxide to the iron complex,
was taken for time
Å, which marks the moment of coordination
and dissociation of the hydrogen peroxide to the iron complex,
was taken for time ![]() , marked by the vertical dashed line.
Before
, marked by the vertical dashed line.
Before ![]() , we see the lines arising from the hydrogen bonded
solvent water molecules to the yet intact hydrogen peroxide and
from the H
, we see the lines arising from the hydrogen bonded
solvent water molecules to the yet intact hydrogen peroxide and
from the H![]() O
O![]() hydrogen H
hydrogen H![]() (distinguished by the bold
line in the graphs). The
(distinguished by the bold
line in the graphs). The
![]() distance,
fluctuating around 1 Å has been left out for clarity. After
distance,
fluctuating around 1 Å has been left out for clarity. After
![]() , we see the lines arising from the hydrogen bonded
solvent water molecules to the, at first, leaving OH. radical
and later formed water molecule oxygen. Note that the change from
OH. radical into water molecule occurs almost 300 fs later
in the initial pathway compared to the relaxed pathway and that the
OH. abstracts H
, we see the lines arising from the hydrogen bonded
solvent water molecules to the, at first, leaving OH. radical
and later formed water molecule oxygen. Note that the change from
OH. radical into water molecule occurs almost 300 fs later
in the initial pathway compared to the relaxed pathway and that the
OH. abstracts H![]() (from the formed Fe
(from the formed Fe![]() -OH
-OH![]() )
in the initial pathway but in the relaxed pathway it abstracts
the hydrogen from one of the hydrogen bonded solvent molecules.
Also, we see in the upper graph of the unrelaxed pathway jumps
from two solvent hydrogens to the OH. radical between
)
in the initial pathway but in the relaxed pathway it abstracts
the hydrogen from one of the hydrogen bonded solvent molecules.
Also, we see in the upper graph of the unrelaxed pathway jumps
from two solvent hydrogens to the OH. radical between ![]() ps,
which however do not make it to a H atom at the water molecule
that is formed; this is ``achieved'' by the H
ps,
which however do not make it to a H atom at the water molecule
that is formed; this is ``achieved'' by the H![]() .
.
The initial reaction pathway was constructed by driving hydrogen
peroxide out of the coordination shell from the
[Fe![]() (H
(H![]() O)
O)![]() (H
(H![]() O
O![]() )]
)]![]() complex, as shown
earlier in figure 7.1 between
complex, as shown
earlier in figure 7.1 between ![]() ps. We thus
started from H
ps. We thus
started from H![]() O
O![]() , separated from the complex, that could
hardly have formed a relaxed solvation shell, which however is expected
to exist for separated reactants in the reactant well. This is seen in the
upper graph of figure 7.6 from the absence of
hydrogen bonds from solvent waters before
, separated from the complex, that could
hardly have formed a relaxed solvation shell, which however is expected
to exist for separated reactants in the reactant well. This is seen in the
upper graph of figure 7.6 from the absence of
hydrogen bonds from solvent waters before ![]() . The lower
graph, of reaction pathway no. 10, on the other hand,
shows two solvent hydrogens with
. The lower
graph, of reaction pathway no. 10, on the other hand,
shows two solvent hydrogens with
![]() distances between
1.5-2.0 Å[216], indicating the adoption of
hydrogen bonds from the solvent network and thus indicating that
relaxation of the solvent structure has occurred around hydrogen peroxide.
Because hydrogen peroxide takes part in the three-dimensional solvent
network via the formed hydrogen bonds between solvent molecules and
H
distances between
1.5-2.0 Å[216], indicating the adoption of
hydrogen bonds from the solvent network and thus indicating that
relaxation of the solvent structure has occurred around hydrogen peroxide.
Because hydrogen peroxide takes part in the three-dimensional solvent
network via the formed hydrogen bonds between solvent molecules and
H![]() O
O![]() before the reaction with iron (which is also illustrated in the
first snapshots in figure 7.5)
the leaving OH. radical can terminate much faster via H-bond
wires in the network, resulting in the lower
lifetime
before the reaction with iron (which is also illustrated in the
first snapshots in figure 7.5)
the leaving OH. radical can terminate much faster via H-bond
wires in the network, resulting in the lower
lifetime
![]() .
.
Have we now found the most likely mechanism for the iron(II) catalyzed
dissociation of hydrogen peroxide? Let us first consider the
alternative to the proposed two-step mechanism, namely
the rebound mechanism.
Of course, whether the two-step mechanism is indeed more
favorable than this alternative can in principle only be
established after generating a very large number of independent
reaction pathways, and compare the probabilities of the
two mechanisms. Our transition path sampling sequences indicate
the rebound mechanism to have lower probability than the two-step
mechanism. This can be rationalized from the following motive. In
the rebound mechanism, the leaving OH. has to abstract the
Fe![]() -OH
-OH![]() hydrogen. From our results, however,
we see that the leaving OH. from the solvated hydrogen
peroxide will jump rapidly along an H-bond wire to termination,
which is much more probable, simply because the
Fe
hydrogen. From our results, however,
we see that the leaving OH. from the solvated hydrogen
peroxide will jump rapidly along an H-bond wire to termination,
which is much more probable, simply because the
Fe![]() -OH
-OH![]() hydrogen is further away
after the O-O lysis than the solvent molecules forming H-bonds.
hydrogen is further away
after the O-O lysis than the solvent molecules forming H-bonds.
The alternative mechanism to the two aforementioned ones (with a ferryl ion as the active intermediate) is Haber and Weiss' free radical mechanism. We have given arguments in references bernd3, bernd4 and franco1 why we believe this mechanism to be unlikely. If we would still want to compare its probability to that of the two-step (ferryl ion producing) mechanism, we should consider that for the Haber and Weiss radical mechanism, the leaving OH. radical has to become a ``free radical'' by jumping along an H-bond wire of a number of solvent waters. The OH. then should become disconnected, possibly by a thermal rotation of one of the involved solvent waters, so that the OH. radical cannot jump back to terminate at the aquairon complex. In our rather small system, the occurrence or non-occurrence of the latter event cannot be adequately tested since an OH. radical that leaves the complex to become a free radical via a short wire of, say, three solvent waters, is already too close to the periodic image of the iron complex at which it can terminate. For reaction path sampling of the Haber and Weiss free radical mechanism, a larger unit cell would be essential.
Let us finally note that the AIMD simulations are not without shortcomings. The box size was chosen rather small in order to make the computations feasible. The OH. radical could therefore jump via a hydrogen bonded wire of three water molecules through the unit cell to a neighboring periodic copy of the cell. So, although the reactants have at least one complete hydration shell, and the major solvation contributions are included in the calculations, the long range effects are approximated and the simulations can, in the future when computers become faster, be improved by increasing the box size and the number of solvent molecules in the box. Other (minor) error sources are the neglect of pressure effects in the NVT ensemble, the classical treatment of the nuclear dynamics neglecting tunneling and zero point energy effects, and the accuracy of electronic structure method which is with the present exchange-correlation functions of DFT about 1 kcal/mol, and significantly larger (due to approximations in the PAW method) when changes in oxidation state of the metal are involved, see section 7.3.