Fenton's reagent, used for oxidation of organic substrates, consists of
an aqueous ferrous ion solution which catalyzes oxidation with hydrogen
peroxide. The active species is believed
to be either the OH. radical produced via reaction
equation 7.1[156],
Usually, Fenton chemistry takes place in aqueous solution and the solvent effects are expected to play an important role in the reaction mechanisms. The static DFT calculations, for example, pointed out that solvent water molecules can act as bridges in the hydrogen transfer occurring in the second step of reaction 7.3 and of course also the OH. radical can jump via solvent water molecules, opening new pathways. The reaction free energy profile of a chemical reaction in aqueous solution can in principle be estimated with ab initio molecular dynamics using an umbrella potential or a geometric constraint (see e.g. refs. bernd2,MeSp98,MeSp298) if a good estimate of the transition state and an appropriate reaction coordinate are known. These are however not known a priori for our system. To obtain some idea of the possible molecular reaction mechanisms in water, we performed illustrative reaction pathways of the reaction between iron(II) and hydrogen peroxide in water.[171,146] One of the pathways shows a new mechanism, and this pathway will be the initial path for the generation of new pathways in the present study.
|
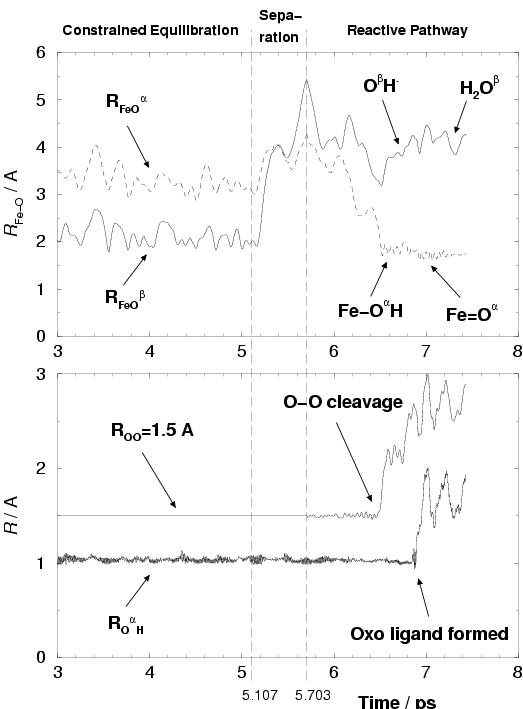
Figure 7.1 shows characteristics of the pathway of
ref. bernd4 that will be used as the initial pathway for the
transition path sampling, we will carry out in the present work.
The relevant geometric parameters (the FeO![]() and FeO
and FeO![]() distances in the upper graph and the hydrogen peroxide
O
distances in the upper graph and the hydrogen peroxide
O![]() O
O![]() and O
and O![]() H distances in the lower graphs,
O
H distances in the lower graphs,
O![]() and O
and O![]() being the H
being the H![]() O
O![]() oxygens with O
oxygens with O![]() the one that becomes connected to iron in the reactive pathway) are
shown as a function of time for this pathway.
The actual reaction pathway starts at a time
the one that becomes connected to iron in the reactive pathway) are
shown as a function of time for this pathway.
The actual reaction pathway starts at a time ![]() ps denoted by one of
the vertical dashed lines. Everything before that time, we will
regard as the preparation for this pathway. The first 5 picoseconds
are merely equilibration of the system, which consists of hydrogen peroxide
coordinated to pentaaqua iron(II) and 26 solvent water molecules.
The cubic unit cell was subject to periodic boundary conditions and
a Nosé thermostat maintained an average temperature of
ps denoted by one of
the vertical dashed lines. Everything before that time, we will
regard as the preparation for this pathway. The first 5 picoseconds
are merely equilibration of the system, which consists of hydrogen peroxide
coordinated to pentaaqua iron(II) and 26 solvent water molecules.
The cubic unit cell was subject to periodic boundary conditions and
a Nosé thermostat maintained an average temperature of ![]() K.
To avoid a premature breakup of the complex, the H
K.
To avoid a premature breakup of the complex, the H![]() O
O![]() oxygen-oxygen bond distance was constrained to 1.5 Å.
At
oxygen-oxygen bond distance was constrained to 1.5 Å.
At ![]() ps, indicated by the first vertical dashed line,
we constrained the five FeO bond distances between the iron ion and
the water ligands and decreased them from the average
ps, indicated by the first vertical dashed line,
we constrained the five FeO bond distances between the iron ion and
the water ligands and decreased them from the average
![]() Å to
Å to
![]() Å in 100 AIMD
steps, which forces
the sixth ligand, H
Å in 100 AIMD
steps, which forces
the sixth ligand, H![]() O
O![]() , out of the coordination shell.
At
, out of the coordination shell.
At ![]() ps, we have arrived at a configuration with pentaaquairon(II)
(with a vacant coordination site) and hydrogen peroxide separated
from each other by a little more than 4 Å with a set of atomic
momenta that will lead to further separation. This configuration we
take as the starting point for our reaction pathway and we reverse
all atomic velocities to obtain a set of momenta that will lead to
approaching reactants. Also, the velocities of the fictitious plane
wave coefficient dynamics are reversed as well as the velocity of
the Nosé thermostat variable.
Here our reaction pathway starts, with now atomic velocities that will
lead to approaching reactants.
ps, we have arrived at a configuration with pentaaquairon(II)
(with a vacant coordination site) and hydrogen peroxide separated
from each other by a little more than 4 Å with a set of atomic
momenta that will lead to further separation. This configuration we
take as the starting point for our reaction pathway and we reverse
all atomic velocities to obtain a set of momenta that will lead to
approaching reactants. Also, the velocities of the fictitious plane
wave coefficient dynamics are reversed as well as the velocity of
the Nosé thermostat variable.
Here our reaction pathway starts, with now atomic velocities that will
lead to approaching reactants.
The simulation was now continued without the bond distance constraints.
Due to this difference with the separation process, the system initially
tracks back onto the trajectory before the velocities were reversed,
but soon diverges from it strongly, finding a new route which leads
to coordination of H![]() O
O![]() to the iron complex.
Indeed, at
to the iron complex.
Indeed, at ![]() ps, the hydrogen peroxide
coordinates again to the iron complex, but bonds now with the other oxygen,
denoted O
ps, the hydrogen peroxide
coordinates again to the iron complex, but bonds now with the other oxygen,
denoted O![]() , to iron (see again figure 7.1).
As soon as H
, to iron (see again figure 7.1).
As soon as H![]() O
O![]() coordinates to the complex, O-O lysis takes place,
and an iron(III)hydroxo moiety is formed together with an OH. radical.
At about
coordinates to the complex, O-O lysis takes place,
and an iron(III)hydroxo moiety is formed together with an OH. radical.
At about ![]() ps, the OH. radical grabs the Fe(III)-OH hydrogen
to form a water molecule and the ferryl ion.
ps, the OH. radical grabs the Fe(III)-OH hydrogen
to form a water molecule and the ferryl ion.
The mechanism followed by this pathway will be referred to as the
rebound mechanism, because the OH. radical that splits off in the
O-O lysis step rebounds to abstract the hydrogen from the hydroxo
ligand. The rebound mechanism differs from pathways in which the
OH. radical shunts, via a hydrogen bond wire of
solvent water molecules, to a terminating water ligand, which was
observed when starting from hydrogen peroxide coordinated to
pentaaqua iron(II) in water[171]. In the latter case, a
dihydroxo iron(IV) species was formed (much as predicted by the DFT
calculations of the complex in vacuo), which transformed via
proton exchange with the solvent into the ferryl ion a few picoseconds later.
We will refer to this mechanism as the two-step mechanism.
The main question that remains concerning the rebound pathway is:
to what extent is the found reaction pathway dictated by the
manipulation at time ![]() ps, and is it thus a representative pathway?
We attempt to answer this question by applying the transition path
sampling method to generate pathways that will have no memory of this
preparation strategy.
ps, and is it thus a representative pathway?
We attempt to answer this question by applying the transition path
sampling method to generate pathways that will have no memory of this
preparation strategy.
![$\displaystyle \left[\rm {Fe}^{\rm {II}}(\rm {H}_2\rm {O})_5(\rm {H}_2\rm {O}_2)\right]^{2+}$](img644.png)
![$\displaystyle \left[\rm {Fe}^{\rm {IV}}(\rm {H}_2\rm {O})_4(\rm {OH})_2\right]^{2+}
+ \rm {H}_2\rm {O}$](img645.png)
![$\displaystyle \left[\rm {Fe}^{\rm {IV}}(\rm {H}_2\rm {O})_4(\rm {OH})_2\right]^{2+}$](img646.png)
![$\displaystyle \left[\rm {Fe}^{\rm {IV}}(\rm {H}_2\rm {O})_5(\rm {O})\right]^{2+}$](img647.png)