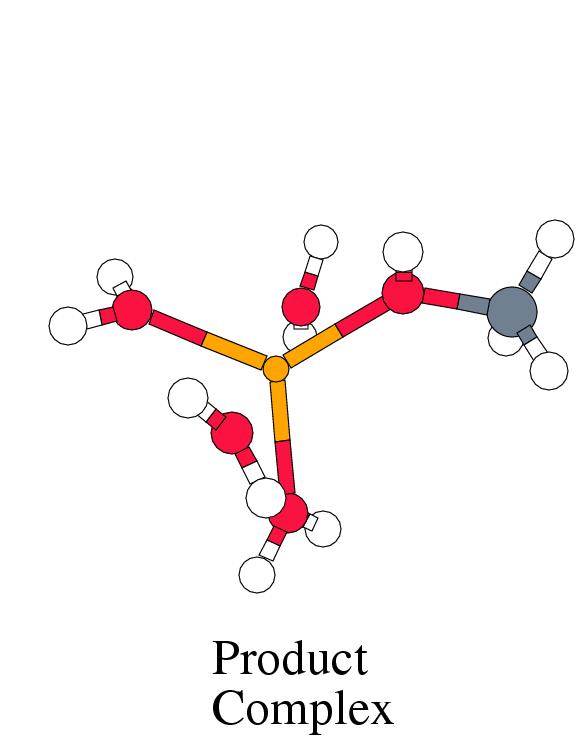
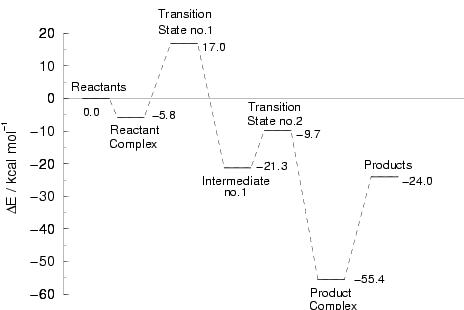
|
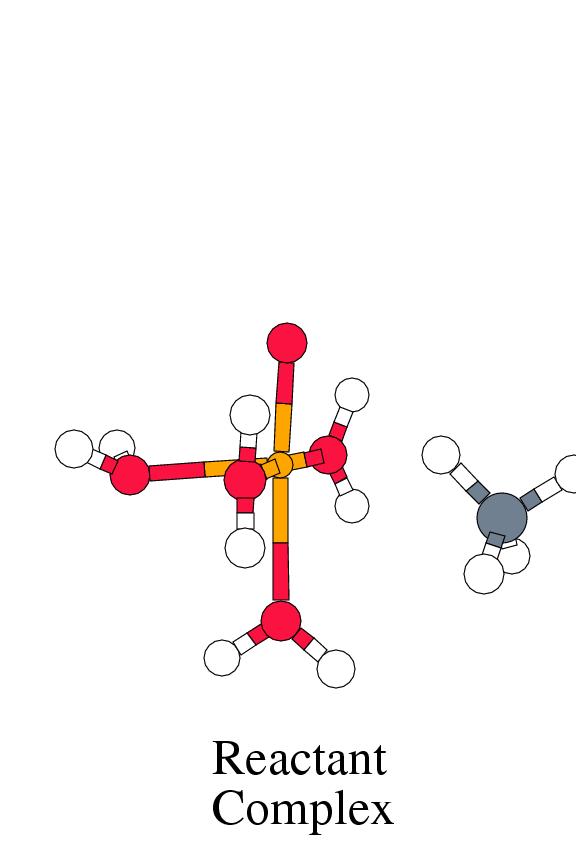
Figure 8.1 shows the energy profile
and the complex geometries along the reaction coordinate
for the reaction between tetraaqua iron(IV) oxo and methane,
following the methane coordination mechanism.
The sum of the energies of the optimized methane molecule and
the optimized tetraaqua iron(IV) oxo complex (i.e. the
reactants), is taken as the off-set for the energy scale.
The initial step is the coordination of methane at the vacant coordination site
of the metal center, forming a weekly bound reactant complex. The interaction
energy is less than 6 kcal/mol. For comparison, the interaction of a fifth
H![]() O ligand at the [Fe
O ligand at the [Fe![]() O(H
O(H![]() O)
O)![]() ]
]![]() complex
equals 29.2 kcal/mol, which indicates that, in aqueous solution,
the required CH
complex
equals 29.2 kcal/mol, which indicates that, in aqueous solution,
the required CH![]() /H
/H![]() O ligand exchange process is endothermic, by more
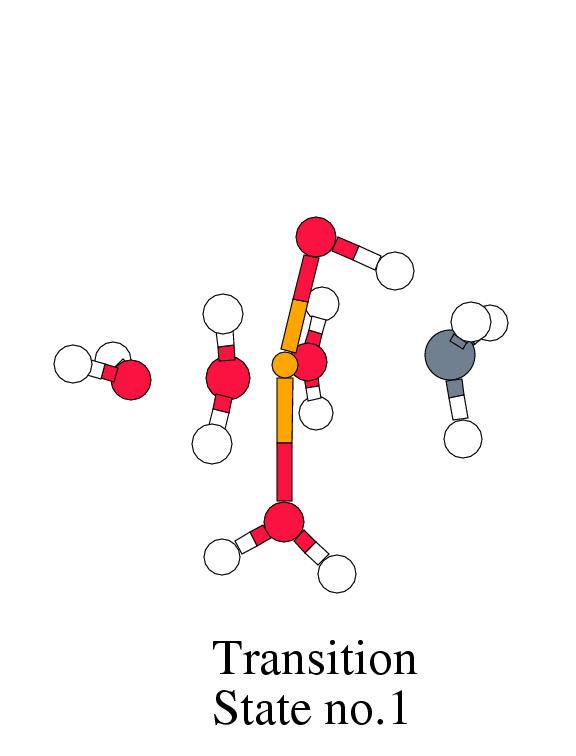
than 20 kcal/mol. The transformation of the reactant complex
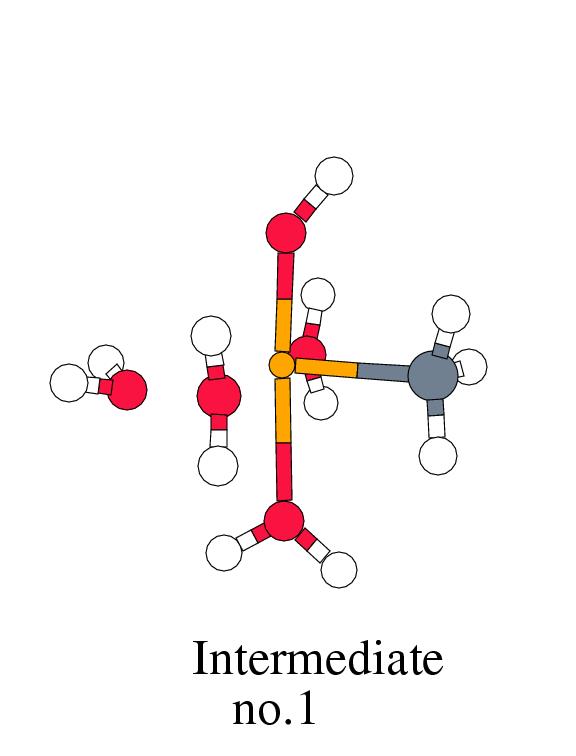
into the product-complex of methanol bound to tetraaqua iron(II) occurs in two
steps. In the first step, a hydrogen is abstracted from the weakly
bound methane by the oxo-ligand, forming a hydroxo ligand and a
(formally) CH
O ligand exchange process is endothermic, by more
than 20 kcal/mol. The transformation of the reactant complex
into the product-complex of methanol bound to tetraaqua iron(II) occurs in two
steps. In the first step, a hydrogen is abstracted from the weakly
bound methane by the oxo-ligand, forming a hydroxo ligand and a
(formally) CH![]() ligand bound to iron(IV). This step is overall exothermic by 15.5
kcal/mol, but has a significant barrier of 22.8 kcal/mol, associated
with a strained four-membered ring in the transition state. In the second
step, the CH
ligand bound to iron(IV). This step is overall exothermic by 15.5
kcal/mol, but has a significant barrier of 22.8 kcal/mol, associated
with a strained four-membered ring in the transition state. In the second
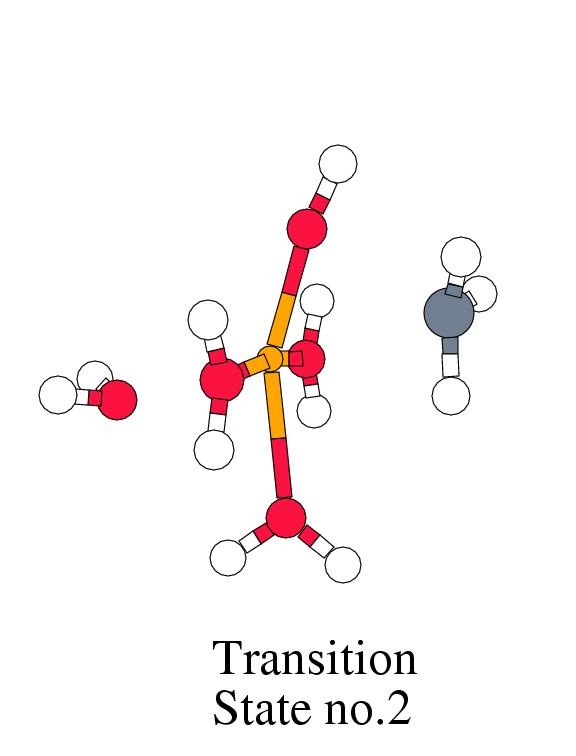
step, the CH![]() group is transferred from the metal center to the
oxygen of the hydroxo ligand, forming a bound methanol group. The
oxidation state of iron is simultaneously lowered from Fe
group is transferred from the metal center to the
oxygen of the hydroxo ligand, forming a bound methanol group. The
oxidation state of iron is simultaneously lowered from Fe![]() to Fe
to Fe![]() , as both ligands (i.e. OH
, as both ligands (i.e. OH![]() and CH
and CH![]() )
donate an electron to the metal
)
donate an electron to the metal ![]() -manifold. The energy barrier for
the, again exothermic, second step is not very high: 11.6 kcal/mol.
The bonding of the methanol to Fe
-manifold. The energy barrier for
the, again exothermic, second step is not very high: 11.6 kcal/mol.
The bonding of the methanol to Fe![]() (H
(H![]() O)
O)![]() is found to be
31.4 kcal/mol strong. The final separation in aqueous solution can however be
expected to be almost thermoneutral (
is found to be
31.4 kcal/mol strong. The final separation in aqueous solution can however be
expected to be almost thermoneutral (
![]() kcal/mol, neglecting solvent
effects) if it is accompanied by the
coordination of a solvent water to the vacant coordination site of the
five-fold coordinated product complex, i.e.:
kcal/mol, neglecting solvent
effects) if it is accompanied by the
coordination of a solvent water to the vacant coordination site of the
five-fold coordinated product complex, i.e.:
|
|
|||||||
| Free | Reactant | Trans. | Inter- | Trans. | Product | Free | |
| Reactants | Complex | State 1 | mediate | State 2 | Complex | Products | |
|
|
|||||||
|
|
1.62 | 1.61 | 1.73 | 1.74 | 1.79 | 2.03 | |
|
|
2.51 | 1.13 | 0.98 | 0.97 | 0.97 | 0.97 | |
|
|
3.02 | 2.20 | 2.06 | 2.46 | 3.18 | ||
|
|
3.62 | 2.34 | 2.70 | 2.18 | 1.48 | 1.43 | |
|
|
1.10 | 1.12 | 1.30 | 2.90 | 2.40 | 2.02 | 1.96 |
|
|
98. | 72. | 90. | 59. | 21. | ||
|
|
|||||||
|
|
1.22 | 1.24 | 1.43 | 1.40 | 1.32 | 1.25 | 1.28 |
|
|
-0.16 | -0.20 | -0.25 | -0.21 | -0.30 | -0.08 | -0.09 |
|
|
0.30 | 0.41 | 0.19 | 0.20 | 0.41 | 0.29 | 0.22 |
|
|
-0.08 | -0.11 | 0.03 | 0.08 | 0.08 | 0.06 | 0.07 |
|
|
|||||||
|
|
3.10 | 3.07 | 3.68 | 3.74 | 3.87 | 3.82 | 3.84 |
|
|
0.73 | 0.72 | 0.28 | 0.37 | 0.26 | 0.04 | 0.00 |
|
|
0.00 | 0.04 | -0.19 | -0.32 | -0.31 | 0.00 | 0.00 |
|
|
0.00 | 0.01 | 0.05 | 0.02 | 0.01 | 0.00 | 0.00 |
Compared to the methane-to-methanol oxidation by bare metal-oxo
species, for which the methane coordination mechanism was found to be the most
likely mechanism, there are a number of differences. In the reactant
complexes of methane coordinated to bare FeO, FeO![]() and FeO
and FeO![]() ,
the binding of methane is
,
the binding of methane is ![]() or
or ![]() -type, with Fe-C
distances as short as 2.32, 2.36 and 2.07 Å
respectively[170]. The interaction energy between CH
-type, with Fe-C
distances as short as 2.32, 2.36 and 2.07 Å
respectively[170]. The interaction energy between CH![]() and
FeO
and
FeO![]() was found to be extremely high, namely 70.3 kcal/mol, compared
to 22.8 and 5.7 kcal/mol, for the high-spin methane FeO
was found to be extremely high, namely 70.3 kcal/mol, compared
to 22.8 and 5.7 kcal/mol, for the high-spin methane FeO![]() and FeO
complexes, respectively. The interaction energy with the MnO
and FeO
complexes, respectively. The interaction energy with the MnO![]() ion (which is isoelectronic with FeO
ion (which is isoelectronic with FeO![]() ) is only 16.2
kcal/mol[191]. Instead, we find for our aqua ligated
complexes only a weak interaction between methane and the iron(IV) complex
(5.8 kcal/mol), with
methane bonded to the metal via a single hydrogen; the Fe-H bond distance
being 2.17 Å, and a Fe-C distance of 3.02 Å. The relevant geometrical
parameters, as well as Mulliken charges
) is only 16.2
kcal/mol[191]. Instead, we find for our aqua ligated
complexes only a weak interaction between methane and the iron(IV) complex
(5.8 kcal/mol), with
methane bonded to the metal via a single hydrogen; the Fe-H bond distance
being 2.17 Å, and a Fe-C distance of 3.02 Å. The relevant geometrical
parameters, as well as Mulliken charges ![]() and spin populations
and spin populations ![]() are
compiled in table 8.2, for the structures
shown in figure 8.1. The Mulliken charge on
iron of
are
compiled in table 8.2, for the structures
shown in figure 8.1. The Mulliken charge on
iron of ![]() Fe=1.22 is relatively high compared to e.g. the
five-fold coordinated [Fe
Fe=1.22 is relatively high compared to e.g. the
five-fold coordinated [Fe![]() (OH)
(OH)![]() (H
(H![]() O)]
O)]![]() H
H![]() O
complex (
O
complex (![]() Fe=0.85[163]), due to the total +2 charge of our
systems, and hardly changes after forming the reactant complex with
methane. Also, the different basis sets used in our work and in that in the
literature can give rise to differences in the Mulliken charges, so that these
comparisons are to be regarded with reservation.
The spin-density on iron(IV) of
Fe=0.85[163]), due to the total +2 charge of our
systems, and hardly changes after forming the reactant complex with
methane. Also, the different basis sets used in our work and in that in the
literature can give rise to differences in the Mulliken charges, so that these
comparisons are to be regarded with reservation.
The spin-density on iron(IV) of ![]() Fe=3.10, on the other hand,
agrees very well that of other iron(IV) complexes, such as the
tetrahydroxo complex (
Fe=3.10, on the other hand,
agrees very well that of other iron(IV) complexes, such as the
tetrahydroxo complex (![]() Fe=3.10[163]) and in MMO models
(
Fe=3.10[163]) and in MMO models
(![]() Fe=3.11-3.55[185,164,186]). The rest of
the spin-density (the total spin-density is 4) is mainly located on the oxo ligand.
The geometries of transition state no.1 (TS1) and the hydroxo intermediate
agree qualitatively with the ones found for this first step for the
bare [FeO-HCH
Fe=3.11-3.55[185,164,186]). The rest of
the spin-density (the total spin-density is 4) is mainly located on the oxo ligand.
The geometries of transition state no.1 (TS1) and the hydroxo intermediate
agree qualitatively with the ones found for this first step for the
bare [FeO-HCH![]() ]
]![]() [170]. Quantitatively, the
bond lengths differ in the order of 0.1 Å, with notably shorter Fe-O
bond lengths of 1.614 Å and 1.675 Å for the TS1 and intermediate,
respectively (where we find 1.73 Å and 1.74 Å),
and a larger O-H bond distance of 1.443 Å for the
transition state (we find 1.13 Å), in the case of the bare
[FeO-HCH
[170]. Quantitatively, the
bond lengths differ in the order of 0.1 Å, with notably shorter Fe-O
bond lengths of 1.614 Å and 1.675 Å for the TS1 and intermediate,
respectively (where we find 1.73 Å and 1.74 Å),
and a larger O-H bond distance of 1.443 Å for the
transition state (we find 1.13 Å), in the case of the bare
[FeO-HCH![]() ]
]![]() chemistry.
The energetics are again very different. For the bare
[FeO-HCH
chemistry.
The energetics are again very different. For the bare
[FeO-HCH![]() ]
]![]() first step, the barrier is only 4.9 kcal/mol (here,
22.8 kcal/mol), whereas the overall transformation energy equals -52.0
kcal/mol (we find -15.5 kcal/mol). The [MnO-CH
first step, the barrier is only 4.9 kcal/mol (here,
22.8 kcal/mol), whereas the overall transformation energy equals -52.0
kcal/mol (we find -15.5 kcal/mol). The [MnO-CH![]() ]
]![]() profile
shows only slightly better comparison, with a barrier of 9.4 kcal/mol and
-36.1 kcal/mol overall.
The CH
profile
shows only slightly better comparison, with a barrier of 9.4 kcal/mol and
-36.1 kcal/mol overall.
The CH![]() ligand interacts much more strongly with the metal center
than CH
ligand interacts much more strongly with the metal center
than CH![]() , giving a bonding energy of -31.2 kcal/mol in the hydroxo intermediate.
The positive Mulliken charge on the carbon in the hydroxo intermediate has decreased
by 0.2 electron with respect to the reactant complex, and gained a spin
density of 0.36. The spin density
on iron of
, giving a bonding energy of -31.2 kcal/mol in the hydroxo intermediate.
The positive Mulliken charge on the carbon in the hydroxo intermediate has decreased
by 0.2 electron with respect to the reactant complex, and gained a spin
density of 0.36. The spin density
on iron of ![]() Fe=3.74 is in between the spins usually found for
Fe(IV) and Fe(III). The second step, the formation of the methanol,
which we found to be exothermic by 34.1 kcal/mol with a moderate
barrier of 11.6 kcal/mol, was found to be energetically unfavorable for the
bare [FeOH-CH
Fe=3.74 is in between the spins usually found for
Fe(IV) and Fe(III). The second step, the formation of the methanol,
which we found to be exothermic by 34.1 kcal/mol with a moderate
barrier of 11.6 kcal/mol, was found to be energetically unfavorable for the
bare [FeOH-CH![]() ]
]![]() case, and was not further discussed[170].
Also with manganese (i.e. bare [MnOH-CH
case, and was not further discussed[170].
Also with manganese (i.e. bare [MnOH-CH![]() ]
]![]() ), this step is not exothermic
(by 0.2 kcal/mol), and has a very high barrier, equal to 35.9 kcal/mol[191].
), this step is not exothermic
(by 0.2 kcal/mol), and has a very high barrier, equal to 35.9 kcal/mol[191].
We conclude that the oxidation of methane to methanol by the tetraaqua
iron(IV) oxo species is exothermic in vacuo following the
methane coordination pathway, with a highest barrier for the hydroxylation step
of 22.8 kcal/mol.
If one were to start from pentaaqua iron(IV)oxo (as in aqueous solution), the
first step is the water-methane ligand exchange reaction, which in vacuo
is endothermic by 23.4 kcal/mol.
The reaction energy profile shows large differences with that of
methane oxidation by a bare FeO![]() ion, which casts doubt on the
use of bare transition metal-oxo species as models for ligated
transition metal oxo moieties, such as in enzymes and in solution.
ion, which casts doubt on the
use of bare transition metal-oxo species as models for ligated
transition metal oxo moieties, such as in enzymes and in solution.