Dr. Ir. Bernd Ensing
Postal address:
Van 't Hoff Institute for Molecular Sciences
University of Amsterdam
PO Box 94157
1090 GD Amsterdam, The Netherlands
Visiting address:
Science Park 904
1098 XH Amsterdam
room: C2.238
phone: (+31) 20 525 5067
email: b.ensing uva.nl
uva.nl

Research projects for Chemistry or Physics Master/Bachelor students
Below are a number of possible research projects for Bachelor and Master students who are interested in performing high-level scientific research using classical and first-principles molecular dynamics simulations of molecular/chemical systems. A significant part of our research involves development of simulation algorithms and software; some projects related to that are found at the bottom for talented students with experience in programming. This list may not be entirely up-to-date, so please feel free to contact me for additional information.
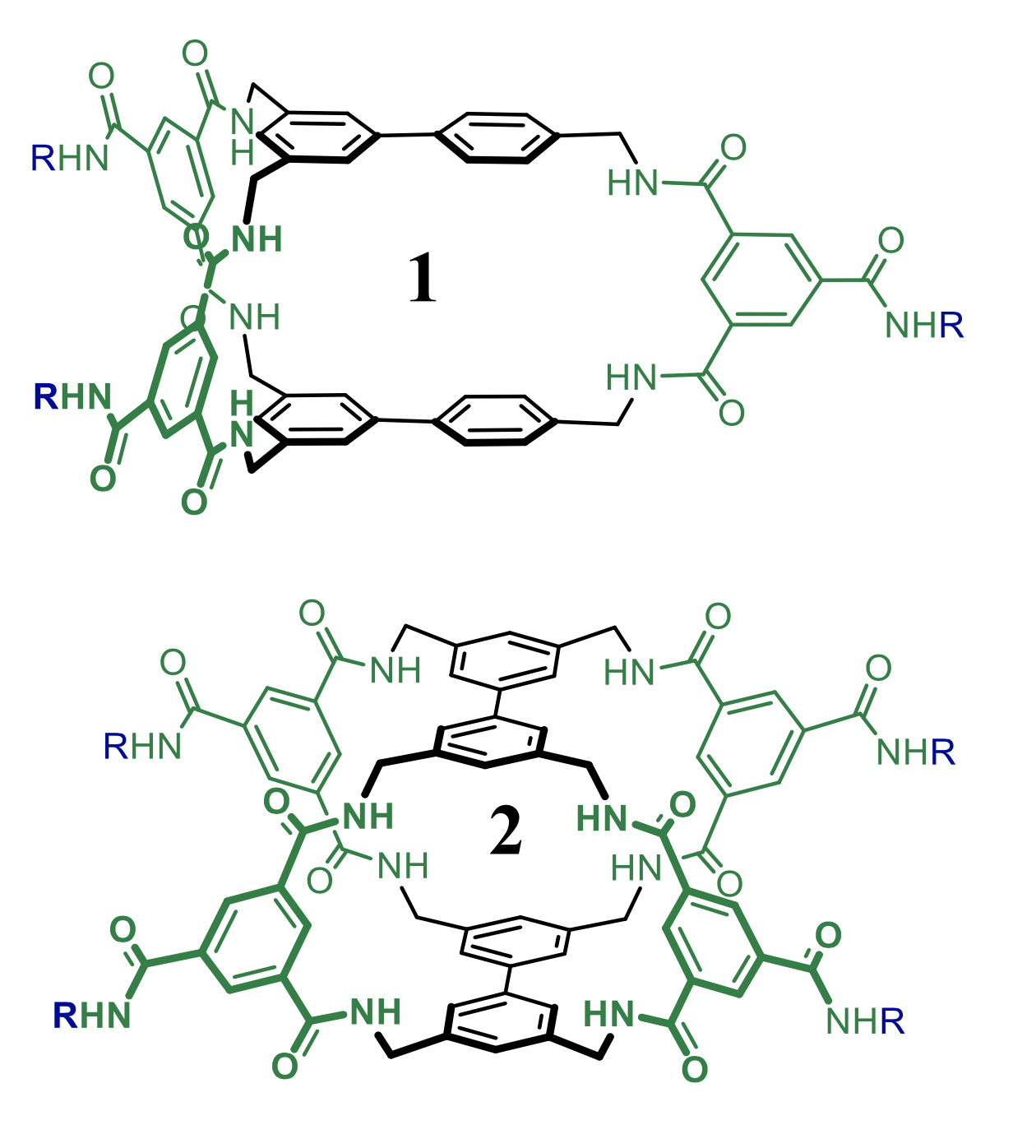
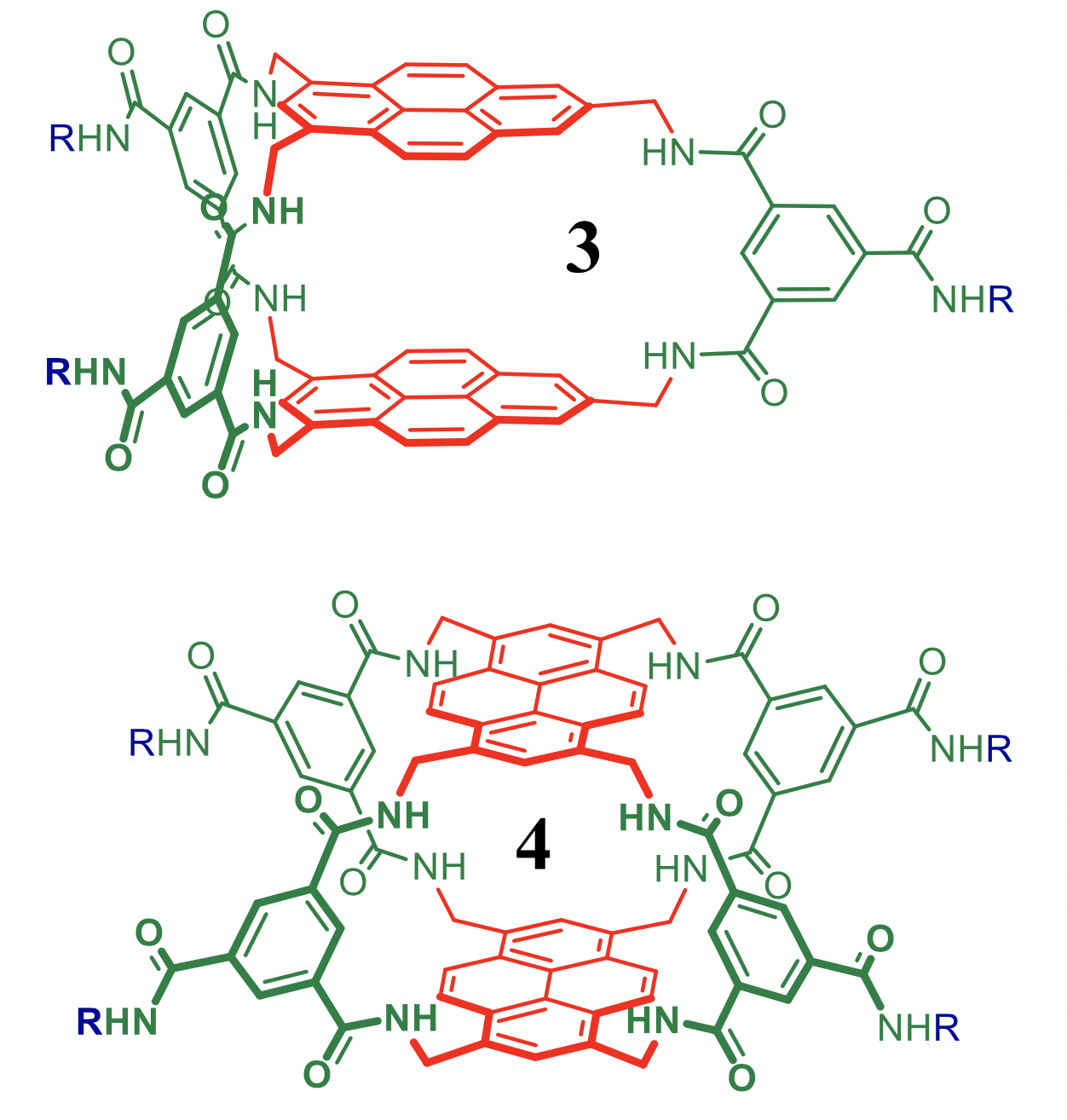
Predicting binding of carbohydrates to artificial receptors
Carbohydrates are Nature’s most versatile class of molecules and fulfil various functions in the immune response system. Selective recognition of a particular (oligo-)saccharide could thus be exploited as an analytical means in biomedical research. Various caging molecules are known to bind carbohydrates in water. We will work on new modular designs with the aim of disclosing artificial receptors with improved recognition properties (affinity and selectivity). The focus of this project is the development of a computational method with which such predictions can be made. The aims include selecting a methodology and calibrating the outcomes to known systems and evaluating a library of potential receptors and predicting which one(s) are the best candidates for experimental exploration. This project is in collaboration with Tiddo Mooibroek of the HomKat group.
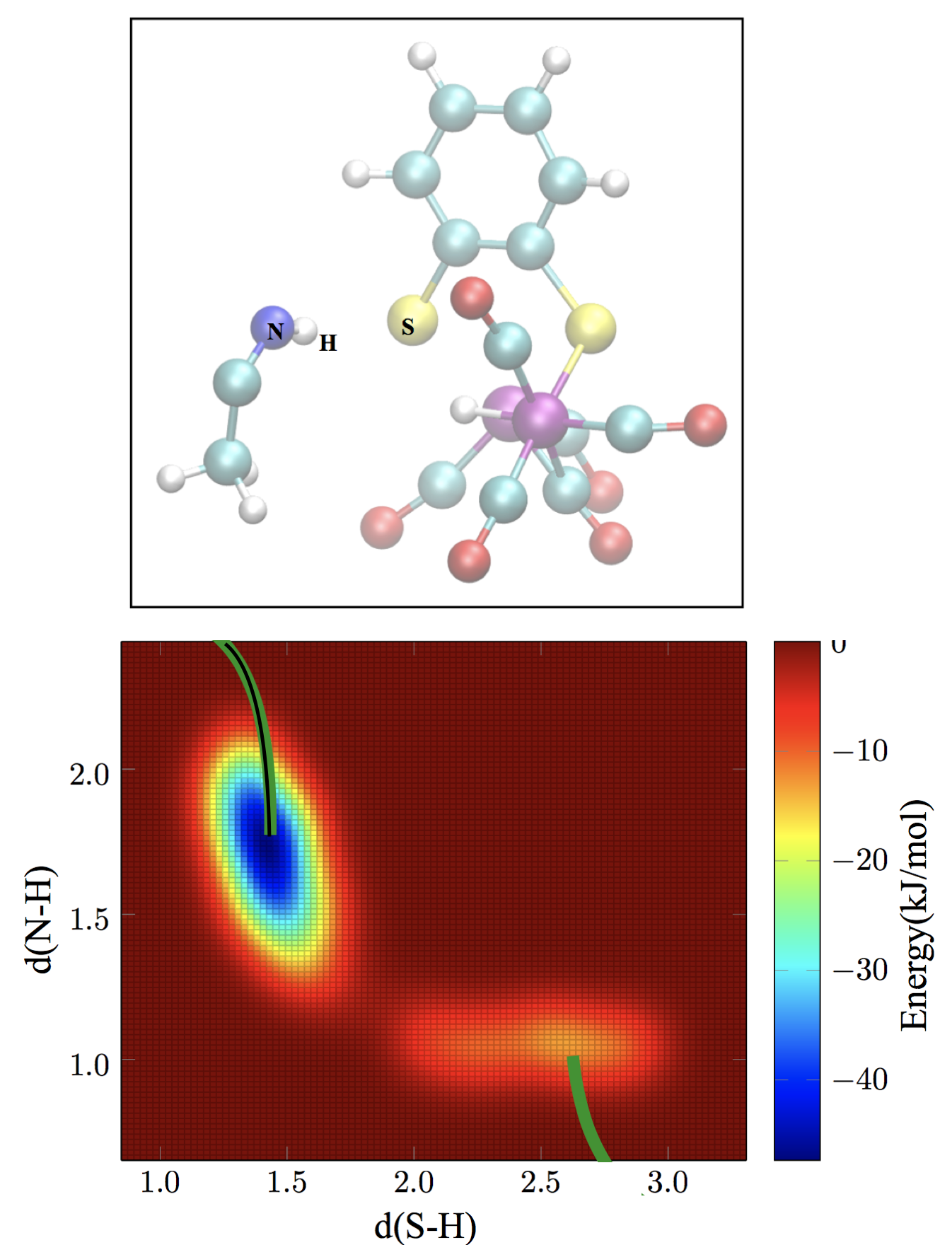
Molecular modeling of homogeneous catalysis
In this project, you will model with DFT calculations and DFT-MD simulations the reaction mechanism of biomimetic di-iron complex catalysed proton reduction to produce molecular hydrogen fuel. This project aims to apply and test our recently developed design rules to find better catalysts. A second research project in this theme concerns mixed QM/MM (DFT/forcefield) MD simulations of the biological hydrogenase enzyme, to uncover why these enzymes are (still) much more active than the artificial mimic-complexes. Both student projects are part of our research line to design better catalysts for sustainable chemistry in collaboration with the homogeneous catalysis group of Joost Reek.
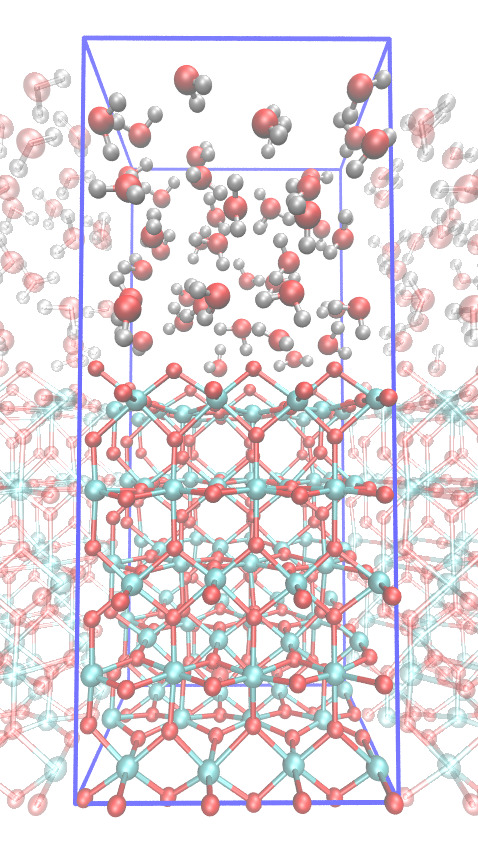
Molecular modeling of heterogeneous catalysis
You will perform DFT-MD simulations of water oxidation reactions catalysed by metal-oxide surfaces in contact with liquid water. The clean metal-oxide surfaces are often very reactive and become quickly "dirty" by hydrolysis, oxidation, and reconstruction. The simulations aim to provide unique insight into the atomic details of the surface, finding surface defects and the reactive sites and obtaining acidity constants, surface energies, and so forth. This student project is part of our research theme on understanding and improving electro-catalysts for clean energy purposes.

Enhanced sampling of electron transfer reactions
The transfer of electrons is a key process in all redox reactions. Recently, we were able to simulate for the first time the spontaneous transfer of an electron from a donor to an acceptor in water solvent using transition path sampling simulations in combination with DFT-MD simulations [Faraday Discuss. 195, 291 (2016)]. Next, we aim to compute the free energy landscape and the rate constants using a different enhanced sampling method, again combined with DFT-MD simulation.
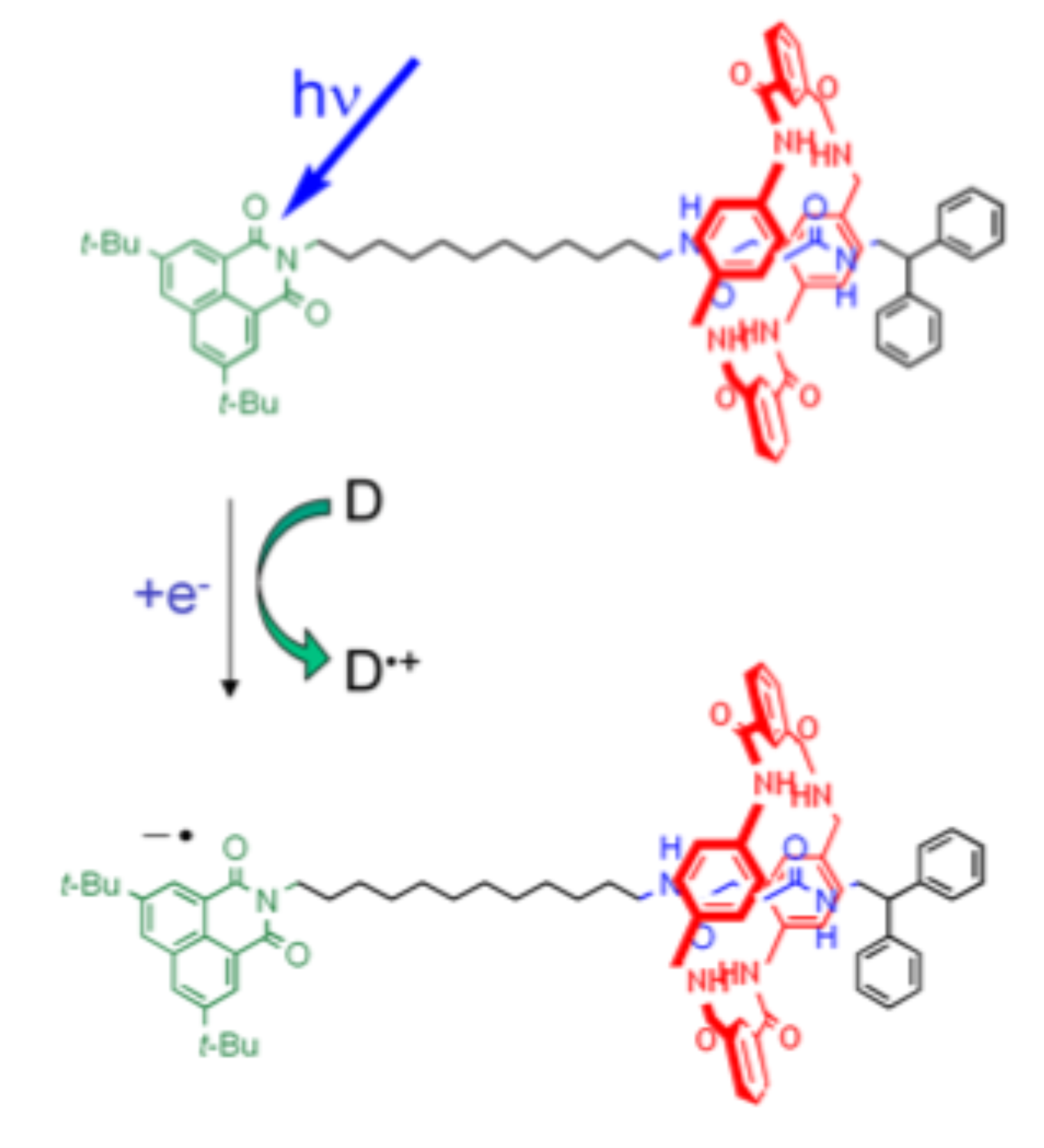
Rotaxane molecular motors
Rotaxane molecules consist of a cyclic molecule surrounding a chain-like molecule, over which it can diffuse from one end to the other. The affinity for the ring to reside at one end or the other can be controlled with (a flash of laser) light, so that rotaxanes can function as molecular switches or molecular motors. We will model the swithing with classical molecular dynamics simulations. One of the aims of the project is to explain why the diffusive mechanism seems to change as a function of the chain length.
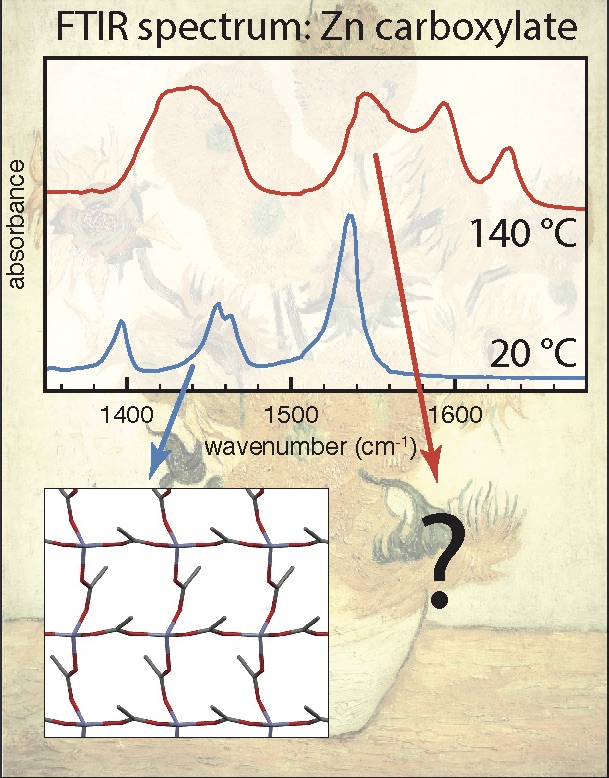
MD for art: Unraveling the structure of liquid-like states of zinc carboxylates
In this project, you will use computational chemistry to investigate the mysterious liquid-like state of so-called 'zinc soaps’, which play a major role in the degradation of old paintings. There is a related experimental BSc project in the Molecular Photonics group. The formation of zinc carboxylates in ageing oil paint leads to paint degradation, often with disastrous consequences. Understanding the structure of these zinc carboxylates is one of the key challenges in art-restoration research. Surprisingly, IR experiments show that zinc carboxylates formed in paintings have a structure that is completely different from the crystalline state. In fact, the state of zinc carboxylates in paintings appears to be similar to the liquid state (which normally occurs only above 130C!). At present the molecular structure of this liquid-like state, both in the pure liquid and in paintings, is still completely unknown. By unraveling this structure using DFT and molecular-dynamics simulations, you will support the development of experiments that help us understand and minimize the degradation of oil paint.
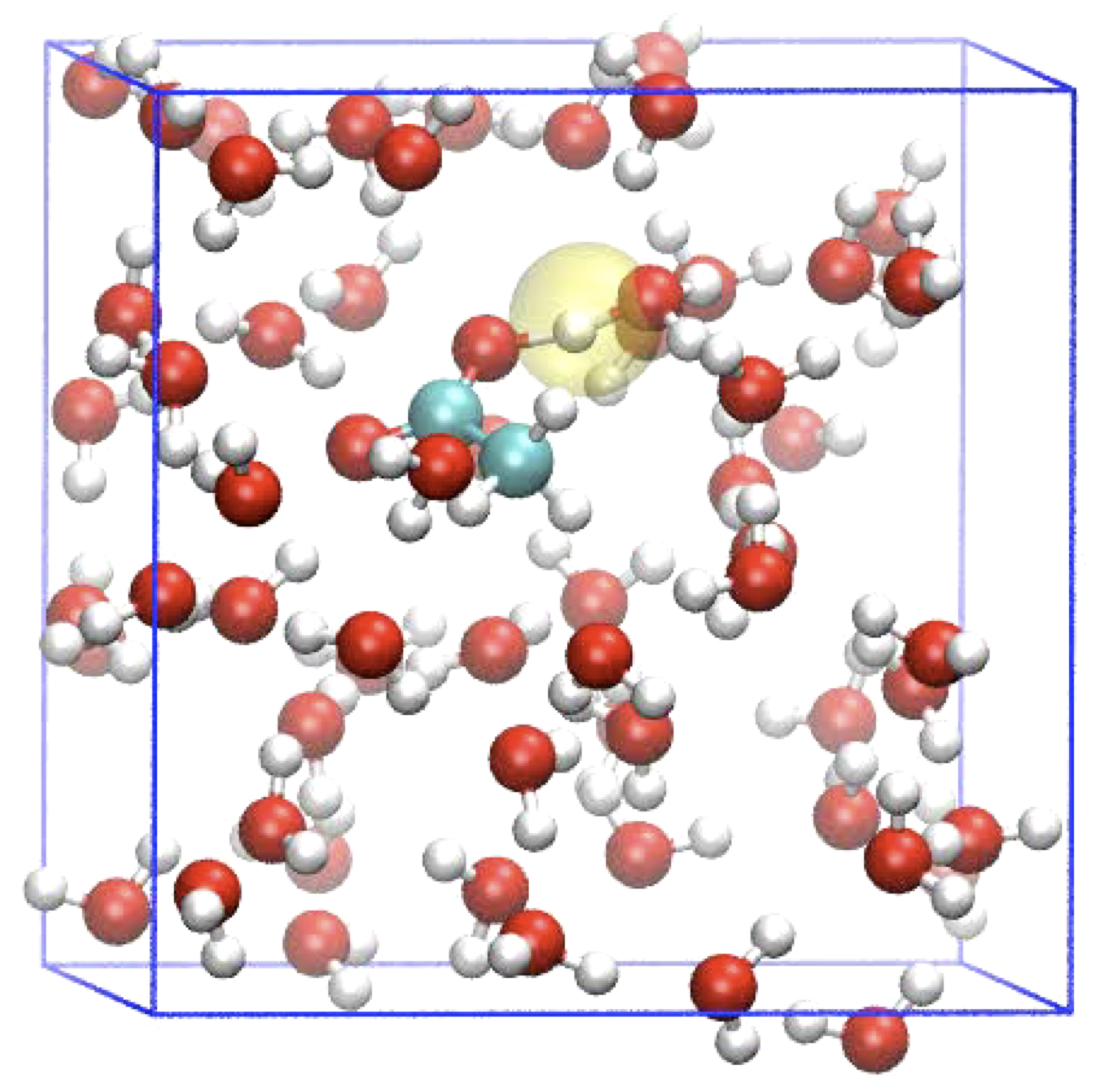
Proton transport
Acid or base catalysed reactivity in aqueous solution involves protons that jump from water molecule to water molecule (aka Grotthuss mechanism). This is difficult to control in a DFT-MD simulation because a reaction coordinate should capture the fact that the moving charge involves every proton jump a different hydrogen. In this project, you will perform free energy simulations, using special reaction coordinates, from which the acidity of molecules and material surfaces can be obtained.
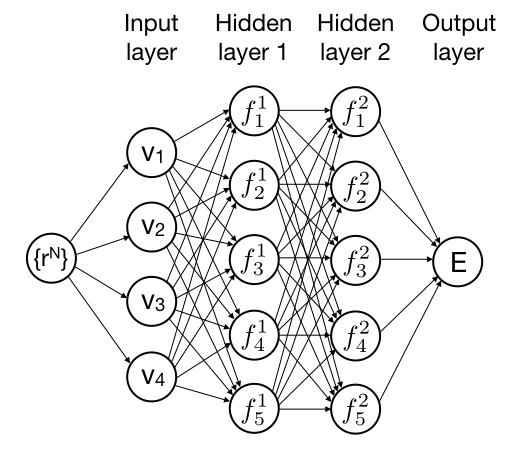
Forcefield simulations based on neural networks
In this project, you will development a simulation program that uses a neural network to describe the interactions between atoms and molecules. By training the neural network with data from accurate quantum chemical (DFT) calculations, the neural network forcefield allows for modeling of chemical reactions, but with much less computational effort than the full-blown DFT-MD simulations. For this project, some experience with programming in C (or C++, Java, Fortran,...) is highly recommended.
Discovering the reaction coordinates of chemical transitions
Most enhanced sampling methods require a detailed description of a reaction coordinate to model a chemical reaction or any other slow, activated, molecular transition. But what if the reaction coordinate is not known? Even partial knowledge of the reaction coordinate is often not enough, leading to hysteresis in computed free energy profiles and large error bars on computed rates and equilibrium constants. In this project, you will develop an analysis tool that takes a molecular dynamics trajectory as input and suggests the relevant "collective variables" that describe the process using normal mode analysis and filtering techniques. For this project, some experience with programming in C (or C++, Java, Fortran,...) is highly recommended.
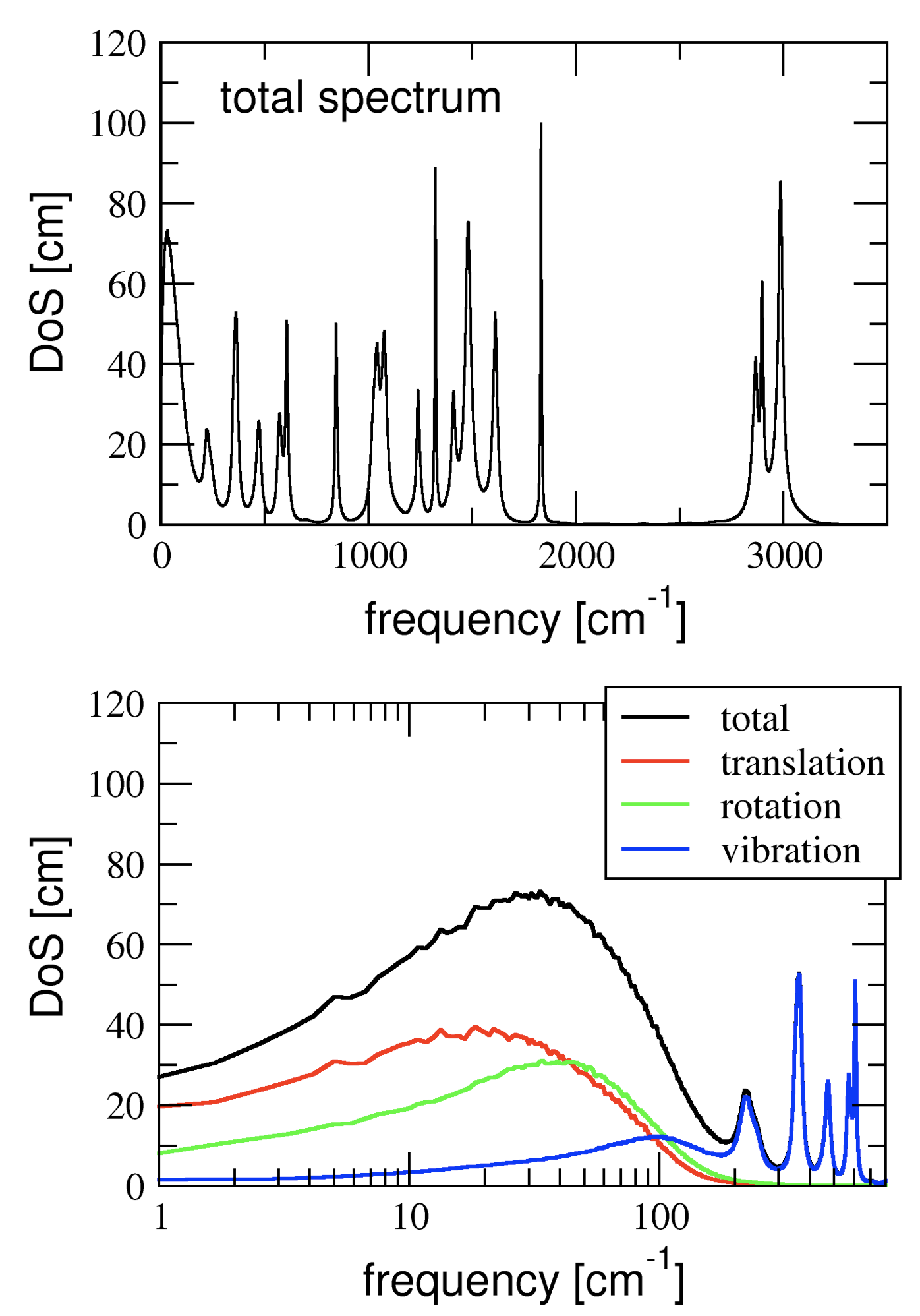
Thermodynamic properties from MD trajectories
DFT-MD simulations use a quantum chemical calculation of the electronic structure to obtain the interactions between the atoms and to propagate the dynamics of the molecules. The dynamics is classical however, and quantum effects on thermodynamic properties computed from the MD trajectories are thus not included. The vibrational spectrum obtained from an MD simulation allows for the quantum corrections within a harmonic approximation. In this project, a tool is developed that computes the vibrational spectrum from an MD trajectory and uses this to calculate various thermodynamic properties, such as the heat capacity and the zero-point-energy. The analysis software is applied to obtain the thermal properties of nanofluids (graphene flakes in organic solvents) in collaboration with the ICN2 Institute for Nano-science and Nano-technology in Barcelona. For this project, some experience with programming in C (or C++, Java, Fortran,...) is highly recommended.